Croissance des microtubules
Contexte biologique
Cette étude se déroule dans le cadre d’une thèse ayant lieu au Francis Crick Institute, London, UK, à propos de protéines qui participent au développement des microtubules en recouvrant leurs extrémités. Ces derniers sont des éléments de cytosquelette impliqués dans l’architecture de la cellule en général et forment le fuseau mitotique lors de la mitose (ou division cellulaire des eucaryotes) en particulier.
L’objectif de l’analyse statistique présentée est de comprendre l’impact d’une de ces protéines sur la croissance des microtubules.
La croissance peut s’effectuer sur l’extrémité (end) plus ou moins du microtubule.
Nous allons voir que cette croissance se fait préférentiellement sur l’extrémité plus.
Chargement des packages
library(tidyverse) # méta-package contenant beaucoup de fonctions utiles pour la manipulation et la visualisation des jeux de données
library(broom) # nettoie les modèles
library(ggpubr) # ggarrange()
library(multcomp) # comparaisons mutiplesImportation des données
Le fichier importé correspond aux données brutes.
jeu <- read_csv("growth.csv")
## Rows: 437 Columns: 8
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: ","
## chr (1): end
## dbl (7): ctrl_buffer, ctrl_NO1, full_length5, C_term5, N_term5, full_length2...
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
jeu
## # A tibble: 437 × 8
## ctrl_buffer ctrl_NO1 full_length5 C_term5 N_term5 full_length2 C_term2 end
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <chr>
## 1 16.4 20.7 30.1 18.8 17.3 20.2 18.4 Plus
## 2 20.6 17.2 19.9 19.8 22.8 21.8 18.3 Plus
## 3 18.3 21.9 23.6 12.6 27.9 18.8 23.9 Plus
## 4 22.3 28.7 20.8 20.6 23.2 23.7 17.7 Plus
## 5 20.5 22.7 21.1 18.8 18.8 16.9 15.6 Plus
## 6 23.6 15.2 23.1 17.4 15.5 18.4 19.4 Plus
## 7 19.2 19.0 21.0 17.0 23.6 20.8 21.2 Plus
## 8 16.7 16.6 19.4 22.7 20.3 16.8 18.3 Plus
## 9 20.3 18.6 14.3 21.4 23.6 18.3 22.9 Plus
## 10 21.5 18.1 17.9 17.4 23.1 18.3 23.6 Plus
## # ℹ 427 more rowsD’un point de vue de statisticienne, il y a en fait 3 variables :
growth_speed, la vitesse de croissance : ce qui est mesurétreatment, le traitement : Control, NO, FL5, FL2, N5, C5 et C2.end, l’extrémité du microtubule : Plus ou Minus.
Le jeu de données est transformé pour qu’il ait 3 colonnes et une ligne par mesure/observation :
jeu.long <- pivot_longer(jeu,
cols = ctrl_buffer:C_term2, # les colonnes à mettre en une seule
names_to = "treatment", # la colonne où vont les noms des colonnes du jeu d'avant
values_to = "growth_speed", # la colonne où vont les mesures
values_drop_na = T # adieu les cases non remplies
) %>%
mutate(treatment = fct_inorder(treatment)) # modalités de treatment rangée dans l'ordre d'apparition
jeu.long
## # A tibble: 1,917 × 3
## end treatment growth_speed
## <chr> <fct> <dbl>
## 1 Plus ctrl_buffer 16.4
## 2 Plus ctrl_NO1 20.7
## 3 Plus full_length5 30.1
## 4 Plus C_term5 18.8
## 5 Plus N_term5 17.3
## 6 Plus full_length2 20.2
## 7 Plus C_term2 18.4
## 8 Plus ctrl_buffer 20.6
## 9 Plus ctrl_NO1 17.2
## 10 Plus full_length5 19.9
## # ℹ 1,907 more rowsVisualisation
Les données brutes sont regardées en premier, car des intervalles de confiance ou des boxplot peuvent cacher des choses.
ggplot(jeu.long) +
aes(x = treatment, y = growth_speed) + # rôle des variables
geom_jitter(width = 0.3, height = 0, alpha = 0.7) + # une observation = un point, avec un bruit horizontal et un peu de transparence (pour les superpositions)
geom_point(stat = "summary"
, fun = "mean"
, colour = "red"
, size = 2
) + # la moyenne est en rouge
facet_wrap(~ end) + # un graphique par End
labs(y = "Growth speed (nm/s)", x = "") + # noms des axes
theme(axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1)) # orientation des labels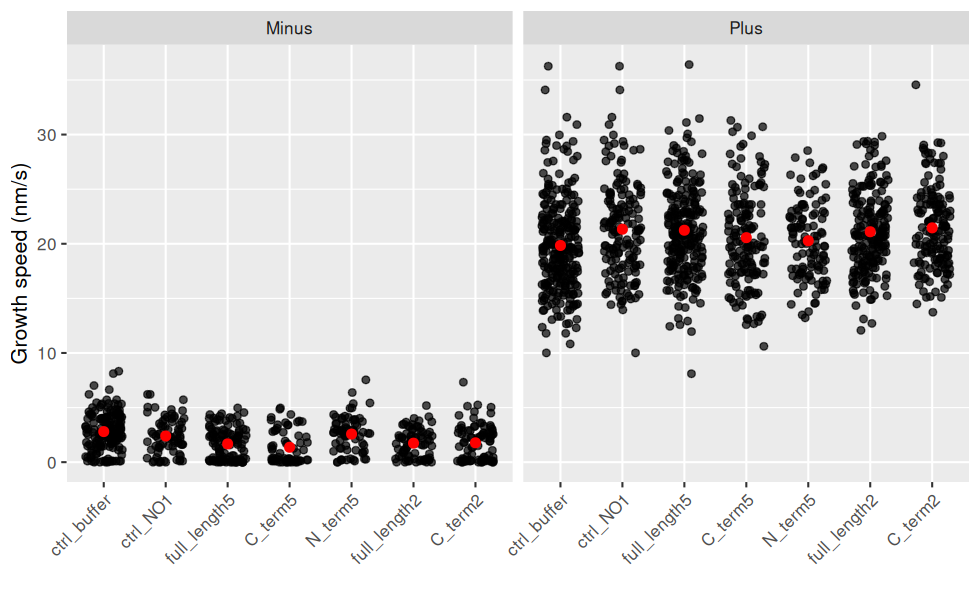
Et avec les intervalle de confiance (bootstrap : aucune hypothèse sur les données sous-jacentes) et les échelles différentes pour les Ends :
ggplot(jeu.long) +
aes(x = treatment, y = growth_speed) + # rôle des variables
geom_pointrange(stat = "summary"
, fun.data = "mean_cl_boot" # bootstrap
) + # le point correspond à la moyenne
facet_wrap(~ end, scales = "free_y") + # un graphique par End
labs(y = "Growth speed (nm/s)", x = "") + # noms des axes
theme(axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1)) # orientation des labels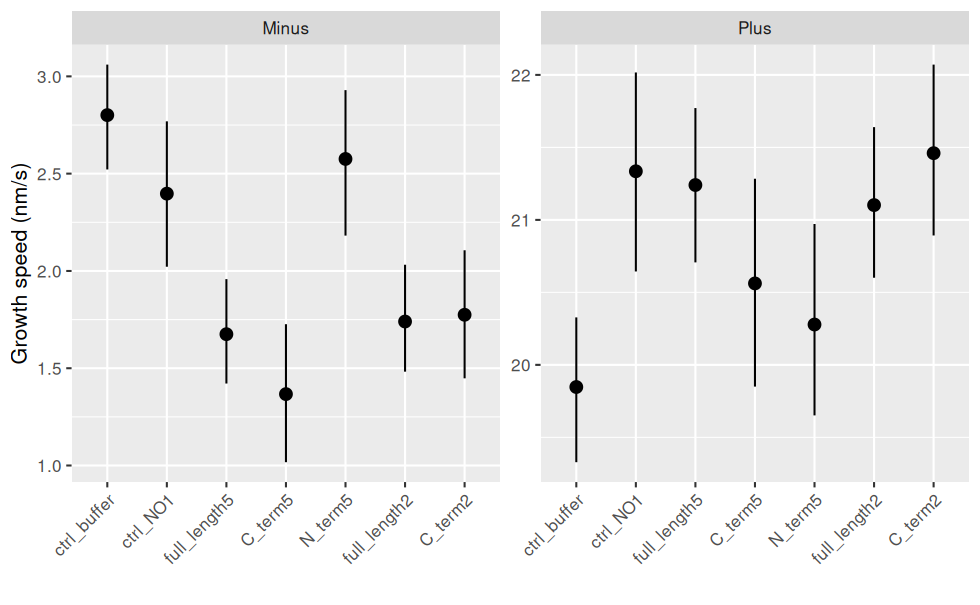
Modélisation
Modèle complet
On suppose une distribution linéaire de la vitesse.
On suppose également l’indépendance des mesures.
On voit que ce n’est pas exact d’ores et déjà, puisque les vitesses faibles (minus) sont minorée par 0.
mod.complet <- lm(growth_speed ~ treatment * end, data = jeu.long)
glance(mod.complet)
## # A tibble: 1 × 12
## r.squared adj.r.squared sigma statistic p.value df logLik AIC BIC
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 0.872 0.871 3.41 998. 0 13 -5064. 10158. 10242.
## # ℹ 3 more variables: deviance <dbl>, df.residual <int>, nobs <int>
tidy(mod.complet)
## # A tibble: 14 × 5
## term estimate std.error statistic p.value
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 (Intercept) 2.80 0.264 10.6 1.26e-25
## 2 treatmentctrl_NO1 -0.404 0.476 -0.848 3.97e- 1
## 3 treatmentfull_length5 -1.13 0.416 -2.70 6.90e- 3
## 4 treatmentC_term5 -1.43 0.496 -2.89 3.86e- 3
## 5 treatmentN_term5 -0.225 0.488 -0.462 6.44e- 1
## 6 treatmentfull_length2 -1.06 0.470 -2.26 2.40e- 2
## 7 treatmentC_term2 -1.03 0.447 -2.29 2.19e- 2
## 8 endPlus 17.0 0.336 50.8 0
## 9 treatmentctrl_NO1:endPlus 1.89 0.586 3.23 1.26e- 3
## 10 treatmentfull_length5:endPlus 2.52 0.517 4.88 1.17e- 6
## 11 treatmentC_term5:endPlus 2.15 0.605 3.55 3.94e- 4
## 12 treatmentN_term5:endPlus 0.656 0.623 1.05 2.92e- 1
## 13 treatmentfull_length2:endPlus 2.32 0.568 4.07 4.82e- 5
## 14 treatmentC_term2:endPlus 2.64 0.566 4.66 3.31e- 6Vérification des hypothèses de normalité sur les résidus :
# Création d'une fonction traçant les ggplots de vérification d'un lm (à la manière de plot.lm())
verifier_lm <- function(modele) {
# résidus standardisés en fonctions des valeurs prédites
hyp1 <- ggplot(modele, aes(.fitted, .stdresid)) +
geom_point() +
geom_hline(yintercept = 0) +
geom_smooth(se = FALSE)
# variance des résidus en fonction des valeurs prédites
hyp2 <- ggplot(modele, aes(.fitted, sqrt(abs(.stdresid)))) +
geom_point() +
geom_smooth(se = FALSE)
# qqplot
hyp3 <- ggplot(modele) +
stat_qq(aes(sample = .stdresid)) +
geom_abline()
# residuals vs. leverage avec les distances de cook
hyp4 <- ggplot(modele, aes(.hat, .stdresid)) +
geom_point(aes(size = .cooksd)) +
geom_smooth(se = FALSE, size = 0.5)
ggarrange(hyp1, hyp2, hyp3, hyp4)
}
verifier_lm(mod.complet)
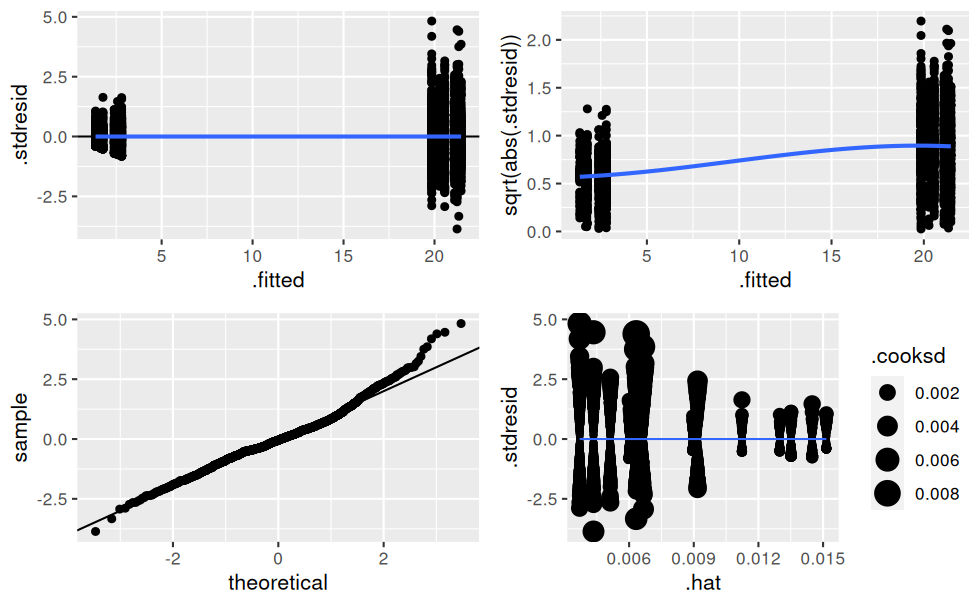
Il y a clairement de l’hétéroscédasticité dans les résidus, c’est-à-dire que leur variance n’est pas constante !
Tranformation de la variable à expliquer Y
Transformation racine carrée de la variable growth_speed, classique en cas d’hétéroscédasticité :
jeu.long <- jeu.long %>%
mutate(tgrowth = sqrt(growth_speed))
mod.t <- lm(tgrowth ~ treatment * end, data = jeu.long)
glance(mod.t)
## # A tibble: 1 × 12
## r.squared adj.r.squared sigma statistic p.value df logLik AIC BIC
## <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 0.898 0.898 0.522 1293. 0 13 -1467. 2965. 3048.
## # ℹ 3 more variables: deviance <dbl>, df.residual <int>, nobs <int>
tidy(mod.t)
## # A tibble: 14 × 5
## term estimate std.error statistic p.value
## <chr> <dbl> <dbl> <dbl> <dbl>
## 1 (Intercept) 1.55 0.0404 38.5 5.94e-240
## 2 treatmentctrl_NO1 -0.145 0.0729 -1.99 4.69e- 2
## 3 treatmentfull_length5 -0.453 0.0638 -7.10 1.76e- 12
## 4 treatmentC_term5 -0.610 0.0759 -8.04 1.58e- 15
## 5 treatmentN_term5 -0.0678 0.0747 -0.907 3.65e- 1
## 6 treatmentfull_length2 -0.374 0.0719 -5.20 2.20e- 7
## 7 treatmentC_term2 -0.417 0.0685 -6.09 1.36e- 9
## 8 endPlus 2.88 0.0514 56.0 0
## 9 treatmentctrl_NO1:endPlus 0.310 0.0897 3.45 5.69e- 4
## 10 treatmentfull_length5:endPlus 0.609 0.0791 7.70 2.14e- 14
## 11 treatmentC_term5:endPlus 0.688 0.0927 7.43 1.68e- 13
## 12 treatmentN_term5:endPlus 0.124 0.0954 1.30 1.93e- 1
## 13 treatmentfull_length2:endPlus 0.520 0.0871 5.97 2.77e- 9
## 14 treatmentC_term2:endPlus 0.602 0.0867 6.95 4.96e- 12verifier_lm(mod.t)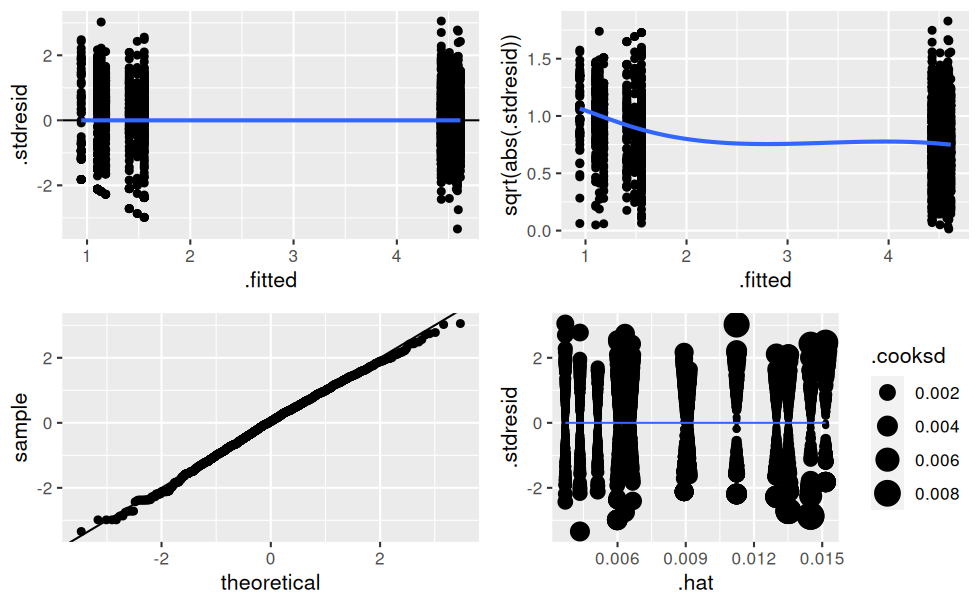
L’AIC et les graphes de vérification des hypothèses sont bien meilleurs maintenant.
Résultats du modèle
On teste les variables explicatives :
anova(mod.t)
## Analysis of Variance Table
##
## Response: tgrowth
## Df Sum Sq Mean Sq F value Pr(>F)
## treatment 6 23.5 3.9 14.341 4.728e-16 ***
## end 1 4527.7 4527.7 16608.383 < 2.2e-16 ***
## treatment:end 6 30.1 5.0 18.390 < 2.2e-16 ***
## Residuals 1903 518.8 0.3
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Les 2 variables treatment et end expliquent growth_speed.
Il y a également interaction : les traitements n’ont pas le même effet que ce soit sur les extrémités plus ou moins.
Comparaison des traitements
Pour faire les comparaisons multiples deux à deux, il ne faut pas d’interaction dans le modèle.
Une nouvelle variable qui combine treatment et end est créée.
jeu.long <- jeu.long %>% mutate(treatend = factor(paste(treatment, end, sep = "/")))
jeu.long
## # A tibble: 1,917 × 5
## end treatment growth_speed tgrowth treatend
## <chr> <fct> <dbl> <dbl> <fct>
## 1 Plus ctrl_buffer 16.4 4.05 ctrl_buffer/Plus
## 2 Plus ctrl_NO1 20.7 4.55 ctrl_NO1/Plus
## 3 Plus full_length5 30.1 5.48 full_length5/Plus
## 4 Plus C_term5 18.8 4.34 C_term5/Plus
## 5 Plus N_term5 17.3 4.16 N_term5/Plus
## 6 Plus full_length2 20.2 4.49 full_length2/Plus
## 7 Plus C_term2 18.4 4.28 C_term2/Plus
## 8 Plus ctrl_buffer 20.6 4.53 ctrl_buffer/Plus
## 9 Plus ctrl_NO1 17.2 4.15 ctrl_NO1/Plus
## 10 Plus full_length5 19.9 4.46 full_length5/Plus
## # ℹ 1,907 more rowsModèle équivalent (sans interaction) :
mod.ssinter <- lm(tgrowth ~ treatend, data = jeu.long)
anova(mod.ssinter)
## Analysis of Variance Table
##
## Response: tgrowth
## Df Sum Sq Mean Sq F value Pr(>F)
## treatend 13 4581.2 352.40 1292.7 < 2.2e-16 ***
## Residuals 1903 518.8 0.27
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Comparaisons multiples 2 à 2 (méthode de Tukey)1 avec une correction de Benjamini-Hocheberg2 pour un risque global alpha de 5% :
comp_treatend <- summary(glht(mod.ssinter, linfct = mcp(treatend = "Tukey")),
test = adjusted("BH")
)
groupes_treatend <- tidy(cld(comp_treatend, level = 0.5)) %>%
separate(1, into = c("treatment", "end"), sep = "/")
groupes_treatend
## # A tibble: 14 × 3
## treatment end letters
## <chr> <chr> <chr>
## 1 C_term2 Minus ab
## 2 C_term2 Plus c
## 3 C_term5 Minus d
## 4 C_term5 Plus e
## 5 N_term5 Minus f
## 6 N_term5 Plus e
## 7 ctrl_NO1 Minus g
## 8 ctrl_NO1 Plus c
## 9 ctrl_buffer Minus h
## 10 ctrl_buffer Plus i
## 11 full_length2 Minus a
## 12 full_length2 Plus c
## 13 full_length5 Minus b
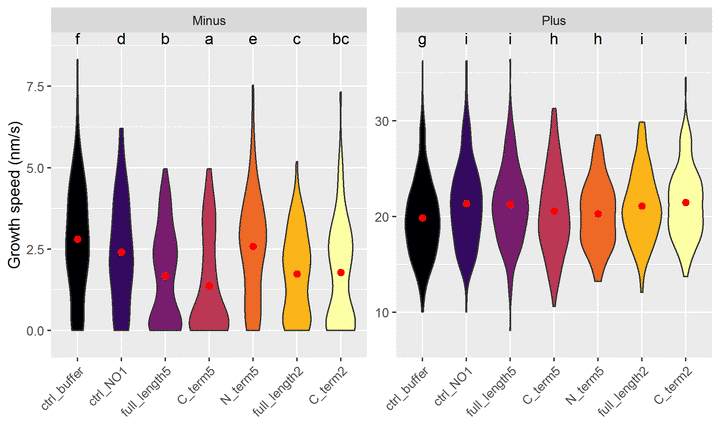
## 14 full_length5 Plus cAjout des groupes sur le graphique :
ggplot(jeu.long) +
aes(x = treatment, y = growth_speed, fill = treatment) +
geom_violin() +
geom_point(stat = "summary"
, fun = "mean"
, colour = "red"
, size = 2
) + # la moyenne est en rouge
geom_text(data = groupes_treatend, mapping = aes(label = .data$letters, x = treatment), y = Inf, vjust = "top") + # ajout des lettres
facet_wrap(~ end, scales = "free_y") + # un graphique par end
labs(y = "Growth speed (nm/s)", x = "") + # noms des axes
theme(axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1), legend.position = "none") + # orientation des labels
scale_y_continuous(expand = expansion(mult = 0.1)) + # élargir l'échelle pour laisser de la place aux lettres
scale_fill_viridis_d(option = "B") # échelle de couleurs distingables par les personnes atteintes de daltonisme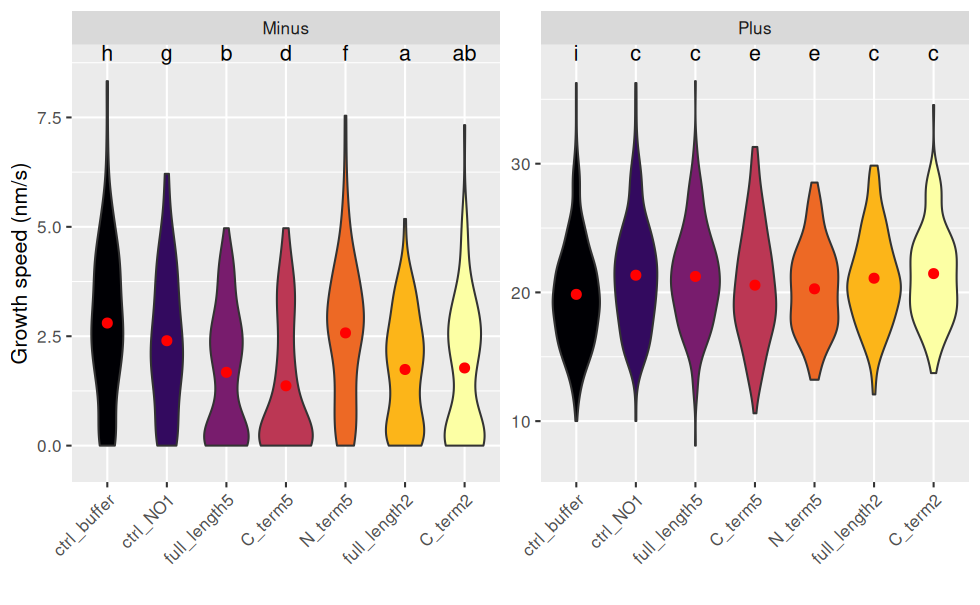
Pour interpréter les lettres : deux combinaisons de traitement/end ayant une lettre en commun ne sont pas statistiquement différentes.
Inversement, s’il n’y a aucune lettre en commun, on peut dire que qu’elles sont statistiquement différentes au risque de 5%.
Exemples :
- Les vitesses de minus sont toutes statistiquement différentes de plus.
- Pour minus : C2 n’est pas différent de FL5 et FL2, sinon tous les autres traitement sont différents 2 à 2.
- Pour plus : Tout le monde est différent du ctrl_buffer ; C5 et N5 ne sont pas différents .
NB : les intervalles de confiance se recoupent, mais ne prennent pas en compte la transformation racine carrée de growth_speed, d’où le choix de représenter un violin plot.
Références
Anna Doizy
Chercheuse, consultante et formatrice freelance
Libre comme l’R
Méthodologie scientifique et analyses de données statistiques